La cirrhose biliaire primitive
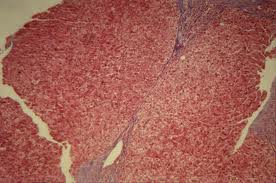 Une place à part a été réservée à la cirrhose biliaire primitive car, dans cette maladie, la cirrhose n’est véritablement constituée qu’à une phase tardive de l’évolution. En réalité, cette affectation réalise une destruction progressive, no- infectieuse, des canaux biliaires intri-hépatiques au niveau interlobulaire. C’es: pourquoi on a pu proposer le synonyme Crocholangite chronique destructive . Nous garderons ici l’expression plus simple de cirrhose biliaire primitive. L’origine de cette cirrhose est inconnue. On a invoqué un mécanisme dysimm. nitaire du type réaction du greffon cont’^ l’hôte.
Une place à part a été réservée à la cirrhose biliaire primitive car, dans cette maladie, la cirrhose n’est véritablement constituée qu’à une phase tardive de l’évolution. En réalité, cette affectation réalise une destruction progressive, no- infectieuse, des canaux biliaires intri-hépatiques au niveau interlobulaire. C’es: pourquoi on a pu proposer le synonyme Crocholangite chronique destructive . Nous garderons ici l’expression plus simple de cirrhose biliaire primitive. L’origine de cette cirrhose est inconnue. On a invoqué un mécanisme dysimm. nitaire du type réaction du greffon cont’^ l’hôte.
L’anatomie pathologique
On décrit quatre stades successifs dans l’évolution histologique de la cirrhose biliaire primitive.
Le stade I
A ce stade, on observe un gonflement et une prolifération des cellules épithéliales des canaux biliaires interlobulaires. Autour de ces canaux se développe un infiltrat inflammatoire dense de cellules mononucléées comprenant parfois en outre quelques éosinophiles. Au sein de ces lésions, on trouve souvent des granulomes plus ou moins bien organisés, parfois véritablement tuberculoïdes, mais sans nécrose centrale. Il s’agit à ce stade de la seule lésion visible, et les espaces portes sont donc par ailleurs normaux. La plaque limitante hépatocytaire est respectée. Ces lésions sont pathognomoni- ques quand elles sont appréhendées par la biopsie hépatique. Ceci n’est malheureusement pas toujours le cas.
Le stade II, ou stade de prolifération ductulaire
A ce stade, les canaux biliaires originels sont remplacés par des néoductules qui n’ont bien souvent qu’une cavité virtuelle. La fibrose, à laquelle s’associe un infiltrat inflammatoire, remplace les canaux biliaires interlobulaires, tandis que les granulomes se font moins fréquents. Cet aspect histologique n’est pas pathognomonique de la maladie, mais il en est hautement évocateur. Des hépatocytes peuvent contenir quelques corps de Mallory.
Le stade III, ou stade de fibrose
A ce stade, l’inflammation régresse, laissant place à des septa fibreux qui s’étendent à partir des espaces portes. Les agrégats lymphoïdes sont toujours présents. Une biopsie hépatique faite alors ne peut être interprétée que comme compatible avec la maladie, sans plus
Le stade IV
Il s’agit du stade de cirrhose, au cours duquel on observe donc de véritables nodules de régénération. Il s’agit d’un stade tardif. A cet état de cirrhose constituée, l’attention peut être attirée vers une cirrhose biliaire primitive par le petit nombre des canaux biliaires et par l’accumulation de lymphocytes. En fait, des chevauchements sont possibles entre les quatre stades histologiques et plusieurs aspects peuvent coexister sur la même biopsie. Au maximum, on peut voir des lésions de stade I au sein d’une cirrhose constituée de stade IV.
La clinique et les examens complémentaires
Les circonstances de découverte
Dans la majorité des cas, il s’agit de femmes d’âge moyen. La prédominance féminine de la maladie est considérable, puisque 90 % des malades sont de sexe féminin. Le début de la maladie est le plus souvent marqué par un prurit isolé. De six mois à deux ans plus tard apparaît un ictère ; dans 25 % des cas environ cependant, l’ictère et le prurit surviennent en même temps. D’autres modes de révélation sont possibles ; en particulier, la cirrhose biliaire primitive peut être découverte devant une augmentation des phosphatases alcalines dans un bilan biologique demandé pour une autre raison. Parfois, une hépatomégalie est fortuitement décelée au cours d’un examen clinique. Rarement, la maladie est révélée par une hémorragie digestive par rupture de varices œsophagiennes. L’hypertension portale est classiquement tardive au cours de la cirrhose biliaire primitive. Cependant, il arrive qu’elle soit au contraire révélatrice.
L’examen clinique
Il existe un ictère ou un subictère. Parfois, le prurit a entraîné des lésions de grattage. Le foie apparaît généralement augmenté de volume dès le premier examen ; il est de consistance ferme, ou même dure, et le bord inférieur peut être tranchant. Assez souvent, on observe des xanthomes cutanés. Il peut s’agir de xanthomes tubéreux, ou encore d’un xanthélasma extensif des paupières supérieures et parfois inférieures. Cette xan- thomatose est en rapport avec l’hyperlipidé- mie, dans certains cas considérable. Une splé- nomégalie est fréquente. Souvent, enfin, on constate des manifestations extra-hépatiques. On peut ainsi observer des arthralgies évoquant au maximum une polyarthrite rhumatoïde ou des manifestations suggérant une dermatomyo- site. Parfois, un syndrome CRST est présent, associant, lorsqu’il est au complet, des calcifications sous-cutanées diffuses, un phénomène de Raynaud, une sclérodactylie et des télangiectasies. L’association sclérodermie et cirrhose biliaire primitive est communément désignée sous le nom de syndrome de Reynolds. Un syndrome sec de Gougerot- Sjôgren, une thyroïdite auto-immune et une glomérulonéphrite constituent les autres manifestations extra-hépatiques possibles de la cirrhose biliaire primitive.
Les examens biologiques
Ils sont le reflet de la cholestase prolongée. La bilirubine est élevée, en fonction de Pictère. Les phosphatases alcalines sont toujours élevées, généralement au-delà de la valeur normale. Au cours des fluctuations de l’ictère, lorsque la bilirubine régresse partiellement, les phosphatases alcalines restent très augmentées. Les IgM sériques sont sélectivement augmentées, et cette anomalie des immunoglobulines est très évocatrice de la maladie. Le cholestérol sérique est souvent élevé, ce qui explique les manifestations xan- thomateuses cutanées ; ses valeurs se situent souvent autour de 4 à 5 g par litre (10 à 13 mmol/litre). Les anticorps anti-mitochondries constituent un signe biologique très important de la maladie. En effet, ils sont ici présents dans 95 % des cas environ, alors qu’on ne les observe que chez 2 à 3 % des malades porteurs d’une cholestase extra-hépatique, et chez aucun témoin sain. On en note aussi l’existence au cours de l’hépatite chronique active auto-immune, et dans ce cas chez 30 % des malades. L’absence d’anticorps anti- mitochondries ne permet donc pas d’éliminer le diagnostic de cirrhose biliaire primitive, mais elle le rend très improbable. La biopsie hépatique fournit des arguments en faveur du diagnostic, parfois patho- gnomoniques, parfois au contraire seulement compatibles avec celui-ci (20). Lorsque le diagnostic de cirrhose biliaire primitive reste incertain, il est fondamental d’éliminer une cholestase extrahépatique. Pour ce faire, on peut parfois recourir à une cholangiographie intravei neuse lorsque le bilirubine n’est pas trop élevée. En fait, on doit le plus souvent faire appel à des moyens plus agressifs d’opacification des voies biliaires, et en l’occurrence à la cholangiographie rétrograde (la cholangiographie trans-hépatique étant a priori moins recom- mandable du fait de l’absence de dilatation des voies biliaires intra-hépatiques).
L’évolution
L’évolution de la cirrhose biliaire primitive est variable, mais généralement sévère. La survie est de 5 à 10 ans en moyenne, mais a tendance à s’allonger actuellement. D’une part, en effet, on dispose d’un traitement efficace dans certains cas, d’autre part, le diagnostic est souvent posé plus tôt. L’ictère connaît des fluctuations ; une mélanodermie s’y ajoute peu à peu, donnant à la peau une coloration jaune-vert. Peu à peu apparaissent une ostéoporose (par malabsorption de la vitamine D), un amaigrissement (du fait de la stéa- torrhée), une hypertension portale (qui, nous l’avons vu, peut être révélatrice) et, finalement, une insuffisance hépato-cellulaire.
Diagnostic différentiel
Le diagnostic différentiel de la cirrhose biliaire primitive comporte les affections cutanées prurigineuses, les cholestases extrahépatiques et les autres cholestases intra- hépatiques.
Les affections cutanées prurigineuses
Les patients sont assez souvent vus, au début, par les dermatologues, en raison du prurit. En réalité, les lésions cutanées, si elles existent, ne sont que des lésions de grattage ; elles sont la conséquence du prurit et non sa cause. Il est nécessaire, devant un prurit sans cause cutanée objective, d’envisager une cholestase et donc de demander un dosage des phosphatases alcalines.
Les cholestases extra-hépatiques
La nécessité de les éliminer justifie l’opacification des voies biliaires au moindre doute, si nécessaire par cholangiographie rétrograde. L’absence de dilatation des voies biliaires intra-hépatiques à l’échographie n’est pas suffisante à cet égard. Ainsi peut-on notamment éliminer un cancer des canaux hépatiques ou une cholan- gite sclérosante.
Les autres cholestases intra-hépatiques
Les autres cholestases intra-hépatiques doivent être éliminées. Il s’agit d’hépatites médicamenteuses et des formes cholestati- ques des hépatites chroniques actives. Certes, l’histologie oriente souvent valablement dans un sens ou dans l’autre. Cependant, il est des cas où l’on ne peut trancher. Il est alors habituel de tenter un test thérapeutique par une corticothérapie. Celle-ci est en effet en général efficace dans les hépatites chroniques cho- lestatiques, alors qu’elle est sans effet dans la cirrhose biliaire primitive. Dans certains cas cependant, ce test thérapeutique laisse lui- même plàner un doute diagnostique. D’autre part, la sarcoïdose réalise dans certains cas une cholestase intra-hépatique, très difficile à différencier de la cirrhose biliaire primitive. Cependant, il existe le plus souvent au cours de la sarcoïdose d’autres localisations granulomateuses qui permettent le diagnostic. Dans ce cas, en outre, les anticorps anti-mitochondries sont négatifs. Le dosage de l’enzyme de conversion de l’angio- tensine fournit un appoint au diagnostic ; généralement élevé en effet au cours de la sar- coïdose, il ne l’est qu’exceptionnellement au cours de la cirrhose biliaire primitive.
Le traitement
Comme c’est le cas pour toutes les maladies dont l’étiologie est mal connue, le traitement de la cirrhose biliaire primitive reste décevant. On sait actuellement que les corticoïdes n’ont pas d’efficacité dans cette maladie, non plus que l’azathioprine. La D-pénicillamine semble de plus d’intérêt. Débuté à la dose de 150 mg par jour,la posologie étant progressivement portée à 600 mg par jour après quelques semaines, ce traitement est capable d’améliorer les paramètres biologiques de la maladie. Surtout, il a été montré plus récemment qu’il est également capable de prolonger la survie des malades. Malheureusement, l’emploi de ce médicament est limité par la possible survenue de complications assez fréquentes et sérieuses (éruptions cutanées et surtout atteintes rénales et médullaires). Lorsque survient l’une de ces complications, il est parfois possible, après arrêt de la D-pénicillamine, de reprendre le traitement sous corticothérapie de couverture. Il semble que doivent bénéficier du traitement par la D-pénicillamine les malades dont l’histologie correspond aux stades III et IV. Il n’est probablement pas raisonnable de commencer un traitement par la D-pénicillamine chez des malades asymptomatiques ayant une biopsie hépatique au stade I ou II. La cyclosporine A semble également active. Son efficacité est actuellement en cours d’étude. Sa toxicité rénale risque de- limiter l’emploi. En dehors de ce traitement à visée étio- pathogénique, il existe des traitemer:.- symptomatiques qui doivent souvent être u: • lisés. La cholestyramine est généralement eff- cace sur le prurit. L’administration par vo e parentérale aes vitamines liposolubles A, D • (non absorbées par voie digestive du fait de ; cholestase) est indispensable. En cas d’ostéo- porose, on peut y associer des perfusior; intra-veineuses de sels de calcium. La malac- sorption lipidique est combattue au moye » d’un régime riche en triglycérides à chaîner moyennes. En cas de rupture de varices œsophagiennes, une anastomose porto-cave peut êt’e indiquée, dans le cadre du traitement ce l’hypertension portale.
Vidéo: LA CIRRHOSE BILIAIRE PRIMITIVE
Vidéo démonstrative pour tout savoir sur: LA CIRRHOSE BILIAIRE PRIMITIVE
