Maladie wilson
la maladie de Wilson est une maladie génétique liée à une accumulation secondaire de cuivre dans l’organisme et se manifestant par des lésions du foie et du système nerveux.
Histoire
La maladie a été décrite par Kinnier Wilson en 1912. Le gène responsable a été identifié dans la fin des années 1980.
Avant les années 1950, il a toujours été fatale. Les premiers traitements apparu à cette époque, dimercaprol première et la pénicillamine en 1956, trientine dans la fin des années 1960. Utilisation de la date de zinc à partir des années 1960.
Dépistage
Lorsque les deux parents d’un enfant sont porteurs de la maladie, il est essentiel de suivre l’enfant avant les premiers symptômes sont visibles, et en général, ces symptômes apparaissent à l’adolescence. Selon de nombreux témoignages de patients et de leurs familles les symptômes de la maladie de 14 ans.
PROPRIETES PHARMACOLOGIQUES
D-pénicillamine est un des médicaments antirhumatismaux à action lente. Elle a la capacité de chélater les métaux lourds, le sérum de cuivre en particulier, ce qui explique son utilisation dans la maladie de Wilson. Il s’agit d’un thioréducteur la rupture des liaisons disulfures de macroglobulines et produit la dépolymérisation des facteurs rhumatoïdes IgM. Il interfère avec la synthèse du collagène.
Après administration orale, 50-70% des D-pénicillamine sont résolus. L’absorption est réduite par les aliments. La concentration plasmatique maximale est atteinte après environ 2 heures. diminue ensuite, d’abord avec une demi-vie d’environ 1 heure et une demi-vie d’environ 5 heures. D-pénicillamine distribue pratiquement tous les tissus du corps.
Après administration orale de D-pénicillamine, plus de 80% est excrété dans les fèces et l’urine de 48 heures, la fraction liée au collagène est lentement éliminé avec une demi-vie de plusieurs jours.
PHARMACEUTIQUE
cellulose microcristalline, mannitol, crospovidone (Kollidon CL), amidon de maïs, gélatine, talc, stéarate de magnésium, silice colloïdale anhydre (Aerosil 200 V), édétate de sodium.
Pelliculage: dioxyde de titane (E171), talc, macrogol 6000, copolymère neutre d’acrylates et méthacrylates (Eudragit NE 30 D), le polysorbate 80 (Tween 80), 30 carmellose sodique CP, siméthicone.
MALADIE DE WILSON
Rare maladie transmise héréditairement dans le foie et les parties du cerveau (noyaux gris centraux) Elle affecte le métabolisme du cuivre.
L’anomalie génétique se situe sur le bras long du chromosome 13 (13q 20-21). (Mutation H1069Q)
Il est transmis comme un mode autosomique récessif (Voir autosomique récessive)
– L’anomalie essentielle est un défaut dans la synthèse d’une protéine plasmatique responsable du transport du cuivre dans l’organisme: la céruloplasmine.
Ainsi, le cuivre créerait un anormale du foie et dans certaines parties du cerveau. Il en résulte une diminution du taux de cuivre en circulation a augmenté de plasma et l’excrétion urinaire du cuivre.
Biologiquement IgD est augmentée (voir immunoglobulines).
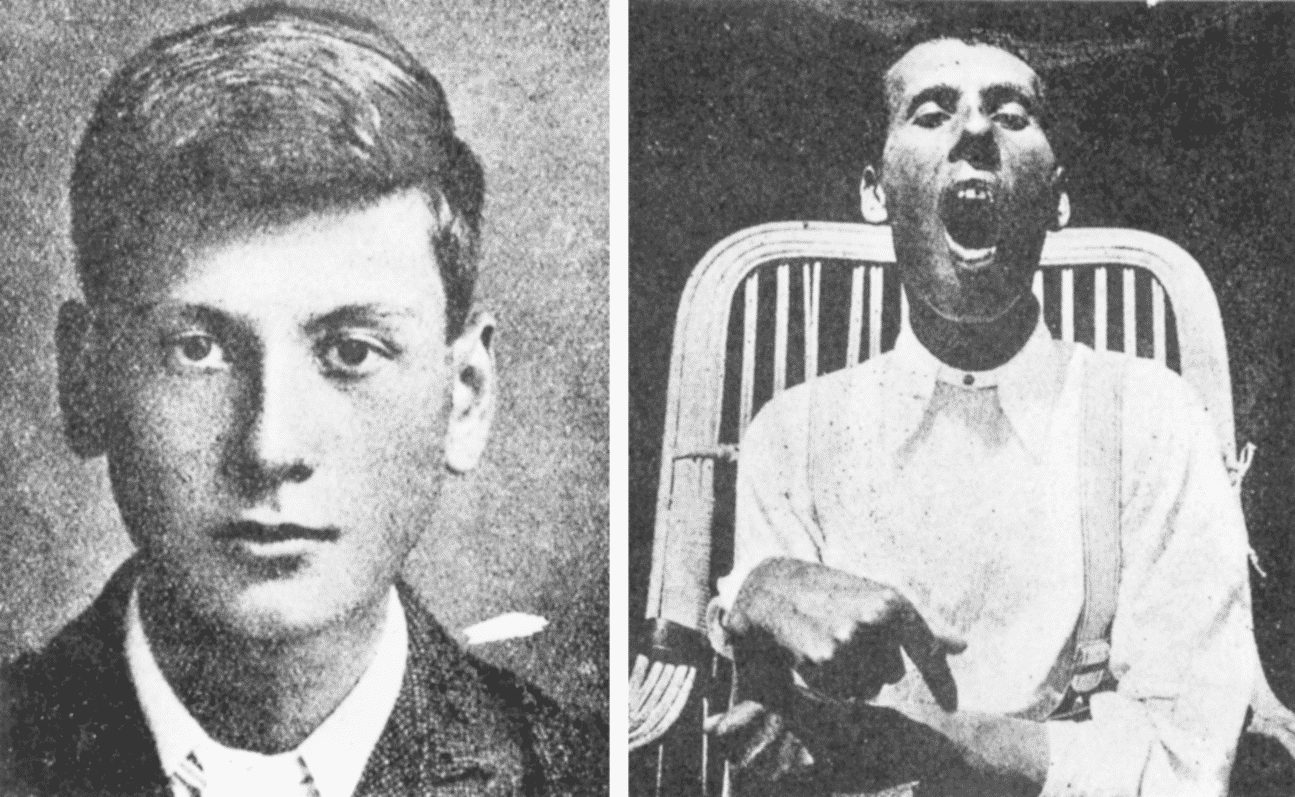
Symptômes:
Les signes commencent à se manifester chez l’adulte par:
– Tremors, en particulier des membres supérieurs
– Rigidité et hypertonie d’action qui détermine les mouvements de la marche et de volontaires
– Une diminution de la motilité et la mobilité
– Les saisies.
– La détérioration des facultés mentales, à un stade ultérieur
– La cirrhose du foie est constante en raison de l’absorption du cuivre sur la glande hépatique.
– Un anneau autour de la cornée verte; cet anneau, qui doit sa couleur à la fixation du cuivre est typique de la maladie et survient généralement au début. (Anneau de Kayser-Fleischer)
EXAMENS DE LABORATOIRE
Le sérum céruloplasmine est diminuée (-cuivre dans les urines accrue (> 100 microgrammes par 24 heures).
-Augmentation des transaminases persistante
– Augmentation de cuivre hépatique
– Le dosage de la céruloplasmine doit être fait entre les membres de la famille afin de détecter les formes précoces de souvent asymptomatique.
TRAITEMENT:
Il doit être institué au début, d’où l’importance d’un diagnostic précoce. Il implique l’utilisation d’un agent chélateur: D pénicillamine qui mobilise le cuivre fixe dans les tissus et favorise son excrétion dans l’urine.
Un chélateur est une substance qui peut se combiner avec un produit toxique (en particulier un métal) pour former un moins toxiques et plus facilement démontable. Ce n’est pas un antidote.
Un second liant peut être utilisé lorsque les effets secondaires de la pénicillamine D semblent
Il s’agit de la Trientine graduellement la dose de 1000 à 1500 mg par jour en deux ou trois repas emporté.
Le traitement consiste à suivre toute une vie en surveillant le nombre de sang et d’urine
Plus le traitement est institué tôt, meilleur est le pronostic à long terme.
Le zinc inhibe l’absorption de la cuisson dans l’intestin
Son utilisation est réservée pour les patients asymptomatiques à la dose de 150 mg par jour en trois doses fractionnées
Il peut être gluconate de zinc sulfate ou en combinaison avec des chélateurs.
La vitamine E doit également être utilisé en combinaison aves chélateurs
Un régime strict n’est pas indispensable, cependant, est d’éviter certains aliments
– Champignons
– Crustacés
– Fruits à coques
– Foie
– Chocolat
– Brocoli
La transplantation hépatique est indiquée lorsque la maladie du foie se développe malgré les médicaments.
formes hétérozygotes (Voir hétérozygotes) asymptomatiques ne doivent pas être traités
Rappeler que le processus doit pas être interrompu brusquement pour éviter une aggravation des délais extrêmement grave de la maladie.
La forme hétérozygote (Voir hétérozygotes) asymptomatiques ne doivent pas être traités.