Maladie de wilson
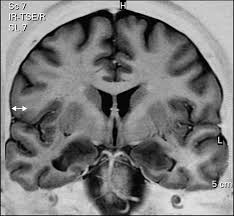
la Maladie de wilson est une maladie génétique liée à une accumulation secondaire de cuivre dans l’organisme et se manifestant par des lésions du foie et du système nerveux.
CLINIQUE
Si les effets thérapeutiques se produire à cette dose, il n’est pas nécessaire de l’augmenter, sinon passer à 900 mg par jour. Être évité autant que possible de dépasser 900 mg par jour.
Après stabilisation de la maladie, réduire la posologie à une dose quotidienne de 600 mg à 900 mg. Ensuite, nous nous en tiendrons à la plus faible dose efficace nécessaire pour obtenir un solde négatif de cuivre. Il est conseillé de ne pas maintenir une dose de 1800 mg par jour pendant plus d’un an.
Allergies · pour les pénicillines et les céphalosporines, en raison de la possibilité, chez certains individus, la Croix-allergie entre les bêta-lactamines et D-pénicillamine.
· Histoire d’accidents graves (aplasie médullaire, pemphigus, myasthénie) dérivés thiols (tiopronine, pyritinol).
· Surveillance étroite cliniques et de laboratoire quand une histoire d’intolérance ou de tiopronine pyritinol.
Fermer les examens cliniques et de laboratoire: hémogramme complet, y compris les plaquettes et protéinurie (toutes les semaines pendant les premiers mois, puis tous les deux mois) éventuellement à la recherche de sang dans les urines, la créatinine sérique.
Pendant le traitement de la maladie de Wilson, il est également nécessaire de surveiller la suppression de la verte anneau de Kayser-Fleischer et l’augmentation du cuivre urinaire, qui doit solde négatif de cuivre urinaire. Aggravation des signes neurologiques dans les premiers mois de traitement formes neurologiques doivent discuter de l’utilité d’un traitement complémentaire.
Lorsque le traitement avec des sels d’or a été arrêté pour effets secondaires, il est, la prudence conseillé de ne pas commencer le traitement par la D-pénicillamine que 6 mois après la disparition de ce dernier.
Les études épidémiologiques sur un nombre limité semblent évoquer un effet réel, mais une malformation très faible de D-pénicillamine par rapport à une population non traitée. Lorsque D-pénicillamine a été donnée dans la maladie de Wilson, aucune anomalie n’a été constatée. L’hypothèse physiopathologique est invoquée désintoxication chélation puis de D-pénicillamine par le cuivre divalent.
Par conséquent, l’utilisation de la D-pénicillamine être envisagée pendant la grossesse que si l’indication est incontestable. Dans le cas de la maladie de Wilson, le traitement ne doit pas être arrêté pendant la grossesse. Dans d’autres cas, une contraception efficace est recommandée.
· Rénale: protéinurie (arrêt en particulier de 1 g par 24 heures). Des cas d’insuffisance rénale sévère ou l’apparition tardive ont été rapportés: syndrome néphrotique, insuffisance rénale aiguë, parfois sévère glomérulonéphrite peut évoluer vers l’insuffisance rénale chronique.
· Du poumon: les cas de pneumopathie interstitielle et oblitérante broncholite ont rarement été rapportés. Ils sont semblables aux complications pulmonaires de la polyarthrite rhumatoïde et peuvent régresser avec l’arrêt du médicament.
· Hématologiques: thrombocytopénie. Cas d’agranulocytose et d’anémie aplasique ont été rapportés.
o biologique: apparition de facteurs antinucléaires, ne nécessitant pas l’arrêt du traitement;
Histoire
La maladie a été décrite par Kinnier Wilson en 1912. Le gène responsable a été identifié dans la fin des années 1980.
Avant les années 1950, il a toujours été fatale. Les premiers traitements apparu à cette époque, dimercaprol première et la pénicillamine en 1956, trientine dans la fin des années 1960. Utilisation de la date de zinc à partir des années 1960.
Diagnostic
La détermination de la teneur en cuivre des tissus du foie est un bon examen (il est augmenté à une maladie de Wilson), mais nécessite l’utilisation de la biopsie hépatique. Dans certains cas, cependant, cette concentration peut être normal (surtout dans les cas de fibrose hépatique avancée).
L’excrétion urinaire est augmentée dans la maladie, mais il ya un certain nombre de faux positifs. Sa mesure peut être sensibilisée chez les enfants en administrant pénicillinamine.
Le test sanguin permet également le diagnostic des formes pré-cliniques, c’est-à-dire avant l’apparition des signes de la maladie qui peut prendre des dizaines d’années à se manifester.
Mécanisme
Elle est due à une anomalie d’un gène impliqué dans le métabolisme du cuivre. la Maladie de wilson est une maladie génétique autosomique récessive: les deux parents sont tout simplement les porteurs de ce gène.
Le gène anormal est situé sur le chromosome 13. 1% de la population générale est porteuse du gène défectueux (hétérozygotes porteurs sains). En France de 1000 à 1500 personnes vivent avec la maladie. Il est principalement exprimé dans le foie et les reins.
Ce gène, appelé ATP7B, code pour une protéine transmembranaire de type ATPase, appelée ATP7B, impliquée dans le transport cellulaire du cuivre intra et extra, pour contrôler la concentration de ce métal et son excrétion dans la bile. Si la protéine est déficiente, le métal s’accumule à l’intérieur des cellules.
Il ya près de 300 mutations décrites de ce gène, mais seulement une poignée de celui-ci est responsable de la plupart des cas de maladie de Wilson. Le type de mutation dépend aussi de l’origine ethnique du patient. En Europe, l’H1069Q type prédomine.
MALADIE DE WILSON
Rare maladie transmise héréditairement dans le foie et les parties du cerveau (noyaux gris centraux) Elle affecte le métabolisme du cuivre.
L’anomalie génétique se situe sur le bras long du chromosome 13 (13q 20-21). (Mutation H1069Q)
Il est transmis comme un mode autosomique récessif (Voir autosomique récessive)
– L’anomalie essentielle est un défaut dans la synthèse d’une protéine plasmatique responsable du transport du cuivre dans l’organisme: la céruloplasmine.
Ainsi, le cuivre créerait un anormale du foie et dans certaines parties du cerveau. Il en résulte une diminution du taux de cuivre en circulation a augmenté de plasma et l’excrétion urinaire du cuivre.
Biologiquement IgD est augmentée (voir immunoglobulines).
Symptômes:
Les signes commencent à se manifester chez l’adulte par:
– Tremors, en particulier des membres supérieurs
– Rigidité et hypertonie d’action qui détermine les mouvements de la marche et de volontaires
– Une diminution de la motilité et la mobilité
– Les saisies.
– La détérioration des facultés mentales, à un stade ultérieur
– La cirrhose du foie est constante en raison de l’absorption du cuivre sur la glande hépatique.
– Un anneau autour de la cornée verte; cet anneau, qui doit sa couleur à la fixation du cuivre est typique de la maladie et survient généralement au début. (Anneau de Kayser-Fleischer)
EXAMENS DE LABORATOIRE
Le sérum céruloplasmine est diminuée (-cuivre dans les urines accrue (> 100 microgrammes par 24 heures).
-Augmentation des transaminases persistante
– Augmentation de cuivre hépatique
– Le dosage de la céruloplasmine doit être fait entre les membres de la famille afin de détecter les formes précoces de souvent asymptomatique.
TRAITEMENT:
Son utilisation est réservée pour les patients asymptomatiques à la dose de 150 mg par jour en trois doses fractionnées
Il peut être gluconate de zinc sulfate ou en combinaison avec des chélateurs.
La vitamine E doit également être utilisé en combinaison aves chélateurs
Un régime strict n’est pas indispensable, cependant, est d’éviter certains aliments
– Champignons
– Crustacés
– Fruits à coques
– Foie
– Chocolat
– Brocoli
La transplantation hépatique est indiquée lorsque la maladie du foie se développe malgré les médicaments.
formes hétérozygotes (Voir hétérozygotes) asymptomatiques ne doivent pas être traités
Rappeler que le processus doit pas être interrompu brusquement pour éviter une aggravation des délais extrêmement grave de la maladie.
La forme hétérozygote (Voir hétérozygotes) asymptomatiques ne doivent pas être traités.