Lupus maladie
Le lupus érythémateux disséminé (LED) est une maladie auto-immune systémique, chronique familiale conjonctifs, c’est-à-dire les tissus conjonctifs multiviscérale, qui se manifeste différemment selon les individus. L’adjectif est associé avec le lupus. Le nom de cette maladie vient de l’aspect classique des lésions cutanées du visage d’un masque de loup (Canis lupus en latin) de Venise (lésion érythémateuse s’étendant du nez sur les joues en forme d’aile de papillon).
mécanisme de la maladie
Le lupus érythémateux disséminé est principalement une maladie auto-immune: des anticorps spécifiques pour les molécules du soi sont produites par le système immunitaire du patient. Ces molécules anormales reconnues comme des émissions étrangères: Ces entités cellulaires sont présentes dans presque toutes les cellules et donc susceptibles d’être détectées lorsqu’une cellule est lysée. Cela conduit à la destruction des cellules environnantes, provoquant des symptômes mentionnés et par la sévérité de la maladie. Les raisons de la nette prédominance féminine de la maladie ne sont pas claires bien qu’il y ait une influence composante hormonale et comme en témoignent les fréquentes épidémies de SLE chez les femmes ménopausées atteintes d’un traitement hormonal substitutif.
Description
SLE est une maladie souvent confondue avec d’autres en raison de sa tendance à «imiter» les symptômes de nombreuses maladies. Il s’agit d’un cas d’école de diagnostic différentiel, en raison de la variété des symptômes de cette maladie et leur tendance à évoluer de façon imprévisible.
critères de classification ACR (American College of Rheumatology)
En utilisant ces critères à un diagnostic de lupus manque de sensibilité et de spécificité dans les premières manifestations de la maladie, ce qui a été évaluée dans plusieurs études de surveillance (spécificité entre 50% et 90% selon les études) <ref > Yokohari R, T. Tsunematsu demande, aux patients japonais, de 1982 révisé American Rheumatism Association critères pour la classification du lupus érythémateux disséminé. Rheum arthrite 1985; 28: 693-8. </ Ref> . En effet, un patient se présente rarement les 4 critères de diagnostic dès le départ et ceux-ci peuvent prendre plusieurs années à apparaître. (Nommé d’après la spécificité de ces études est d’environ 96%).
Diagnostic différentiel
mais aussi avec d’autres auto-immunes conjonctif: la sclérodermie, le syndrome de Gougerot-Sjögren Goujerot-etc.
Étiologie (les causes et les facteurs contributifs)
Certains médicaments (procaïnamide, quinidine, hydralazine) syndromes lupus peut fournir est limité à des lésions articulaires et la peau.
LUPUS ERYTHEMATEUX DISSEMINE
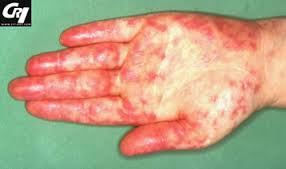
Il s’agit d’une maladie auto-immune systémique (Atteinte des organes multiples) de cause incertaine Il s’agit de la production d’autoanticorps nombreux, et pourtant méconnu, qui est responsable de lésions tissulaires. C’est une maladie du collagène, qui est la substance fondamentale du tissu conjonctif Cette maladie a une prévalence plus élevée parmi les populations noires et asiatiques que dans la population blanche De même, la maladie semble plus grave dans ces populations: Par exemple, les sujets noirs ont un risque plus élevé de développer une néphropathie évolution de plus grave et plus rapide. Les formes juvéniles (qui sont souvent plus sévères) semblent également plus fréquemment dans ces populations. facteurs immunogénétiques ont une grande importance dans la prévalence et les aspects cliniques de cette maladie: la prévalence du tissu groupes HLA B8 et B 15 dans la population mixte de Antilles françaises par exemple Mais aussi DR2 et HLA HLADr3 et chez les sujets présentant un déficit COMPLÉMENT (Fraction C2 ou C4). Un nouveau gène de susceptibilité a été indentifie Ces TNFSF4, l’un de ses haplotypes (qv), plus l’expression TNFSF4 est associée à un risque accru de lupus (1q25) Au début de 2008, les gènes de susceptibilité supplémentaires ont été confirmés HLA – DRB1 sur le chromosome 6 (6p21) ET-FCGR3A FCGR2A sur le chromosome 1 (1q23) – PDCD1 sur le chromosome 2 (2q37) – IFR5 sur le chromosome 7 (7q32) -PTPN22 sur le chromosome 1 (1p13) – STAT 4 ITGAM-sur le chromosome 16 (16p11.2) – KIAA1542 sur le chromosome 11 (11p15.5) -PXK sur le chromosome 3 (3p14.3) – Rs10798269 sur le chromosome 1 (1q25 1) Mais aussi DR2 et HLA HLADr3 et chez les sujets présentant un déficit du complément (qv) (Fraction C2 ou C4). La maladie présente des similitudes avec la polyarthrite rhumatoïde et la sclérose systémique (voir ces termes). Symptômes: L’altération de l’état général avec fièvre est fréquente. Les manifestations cutanées sont les suivants: – Lupus érythème, qui se caractérise par l’apparition d’une éruption rouge foncé sur la face de chaque côté du nez, l’aile d’un papillon ou un carnaval loup (d’où le nom de lupus). Cette éruption peut se produire sur les mains, sur les doigts dorsale. à ce niveau, les plus évocateurs sont vascularite en éruption purpurique péri-type parfois ungéal ou pulpe des doigts peut évoluer vers la nécrose. La réalisation des éminences thénar et hypothénar (voir ces termes) de l’érythème vermillon est caractéristique. D’autres manifestations cutanées sont le type de vascularite Livedo réticulaire- purpura pétéchial, infiltré ou ecchymoses zones érythémato-aspect-violet de frottement et les genoux à Oudes – Les rebelles des lésions urticariennes sans démangeaison – Photosensibilité de la peau retrouve dans environ 30% des cas. – Une perte de cheveux plus ou moins important. – Modifications dans les muqueuses. (Ulcères). Les manifestations articulaires sont précoces et constants, parfois simple douleur commune, parfois avec l’arthrite rhumatoïde de type réel (qv). Elles sont majoritaires dans les mains, les poignets, les genoux. Fondamentalement il n’ya pas de commune destructiion, mais souvent la destruction ou d’un ligament capsulaire. ostéonécrose aseptique peut se produire. (Tête du fémur, l’humérus, ou même les condyles fémoraux. Les manifestations viscérales sont le plus souvent: – Cardiovasculaire (endocardite ou péricardite, myocardite (voir ces termes), phlébite, hypertension, artérite des gros troncs. L’endocardite Libman-Sacks (qv) est devenu dix fois moins fréquent depuis l’utilisation de corticostéroïdes. On peut faire compliqué d’endocardite Osler (voir notes biographiques) – Rein (néphrose Lipoid, rénale chronique), mais aussi lésions glomérulaires minimes, une membrane externe glomérulonéphrite, une glomérulonéphrite segmentaire et focale et diffuse proliférative glomérulo-néphrite dont le pronostic est sombre. L’évolution est poussé par le rein, qui est la principale complication à craindre. Il est donc nécessaire d’enquêter sur l’apparition de cette néphropathie: en recherchant les 24 heures urinaire d’albumine, un compte d’Addis, la créatinine, et la détermination de l’urée sanguine. Cette tendance peut être pendant de nombreuses années, une douzaine en moyenne. – Pleurale (voir pleurésie), fibrose pulmonaire interstitielle diffuse plus ou moins symptomatique. – Peut être responsable de perforation gastro-intestinale ou de saignement dans les voies digestives. colite ulcéreuse, une ascite et une péritonite. – Ganglion, -Hépatomégalie, splénomégalie, – Les yeux qui peuvent couvrir toutes les structures oculaires, dysorique rétinite parfois avec des hémorragies rétiniennes. – Nerve (épilepsie, les troubles mentaux, paralysie faciale voir ces termes). -La baisse de neuro-psychiatriques et cognitifs sont relativement fréquentes. (Convulsions, des maux de tête, confusion, dépression, psychose, troubles cognitifs, focale infarctus cérébral) EXAMENS DE LABORATOIRE D’un point de vue biologique, il ya eu une légère anémie, leucopénie (faible nombre de globules blancs), diminution des plaquettes, vitesse de sédimentation, très rapide, surtout dans le sang et la moelle osseuse, la présence d’une cellule particulière appelée Hargraves cellule. (Maintenant cette recherche a la même importance qu’auparavant puisque l’on peut détecter des anticorps spécifiques à la maladie). Il ya une augmentation de la gamma-globulines sériques. Les anticorps antinucléaires est positive. Il semble que les années de la présence de plusieurs anticorps avant que les symptômes apparaissent. dans l’ordre, les premiers anticorps qui apparaissent sont des anticorps nucléaires (antiphospholipides, anti-Ro, anti-SS-B (LA), environ trois ans avant l’apparition des symptômes et les anticorps anti-ADN double brin, environ deux ans avant et anti-Sm et enfin les antiribonucléoproteine nucléaire. Environ un an avant que les symptômes. – Les anti-ADN natif sont présents dans 70-90% des lupus, à tout moment de la maladie. – Les anticorps anti-histone organismes sont présents dans la même proportion, mais aussi dans les lupus induits. – La présence d’anticorps anti-histone sans la présence d’anticorps anti-ADN double brin est un lupus induit. – Les anticorps spécifiques aux antigènes nucléaires peuvent être hautement spécifique de la maladie comprennent; Anti-Sm, anti-SS-A (Ro), anti-SS-B (LA), anti-UI-RNP. Ce dernier, en l’absence d’anti-ADN double brin, est un marqueur de lupus Bénin sans insuffisance rénale sévère. – Anticorps antiribosomes semble plus spécifique à la dépression en faisant le lupus cérébral. . recherche des anticorps antiphospholipides et la cryoglobulinémie (voir Cryoglobulinémie) peut avoir une valeur pronostique pour la surveillance progression de la maladie La réaction de Waaler-Rose (qv) et le test au latex est positif. TRAITEMENT: Sera utilisée: – Les médicaments anti-inflammatoires en cas d’arthralgies mineurs ou de poussée bénigne – Les médicaments antipaludiques (fundus de contrôle doit être régulier) – Corticoïdes, – Des médicaments immunosuppresseurs. – En cas de résistance au traitement peut être utilisé cyclophosphamide ou azathioprine (en particulier dans les cas graves dommages causés aux reins). Thalidomide (en particulier dans les formes très graves de la peau). Il semble que l’administration de DHEA, sans interrompre le processus actuel, améliore les symptômes de la maladie: Des éclosions ont été moins nombreux et plus rémissions prolongées. DHEA (qv) est une hormone similaire (ce qui est un précurseur) des anomalies œstrogènes et androgènes dans le lupus et qui pourrait expl
iquer l’utilité de cette thérapie complémentaire dans cette maladie. Enfin, dans certains cas autologue de moelle osseuse pourrait être mis en œuvre. (Sévère réfractaire). Dans les cas graves, la plasmaphérèse> (qv) peut être envisagée. Si RENAL (qv) sévère, l’hémodialyse et la transplantation rénale (voir ces termes) peuvent également être considérés. Ce traitement peut prolonger la rémission. L’espoir repose sur l’utilisation thérapeutique des anticorps monoclonaux bloquant (ligand CD40) et l’administration de peptides qui peuvent induire des états de tolérance (peptide anti-B anti peptide-T.