Atteintes auditives de l'oreille interne:Atteintes congénitales et héréditaires
 Les hypoacousies de perception existant dès la naissance sont des atteintes congénitales. Certaines, mais pas toutes, sont héréditaires, c’est-à-dire génétiques, et les autres sont la conséquence de complications de la grossesse. La plupart sont dues à des lésions de la cochlée. Il peut y avoir destruction des structures vasculaires membraneuses qui soutiennent l’organe de Corti (malformation de Scheibe) ou perte d’éléments nerveux en partant des cellules ciliées et en allant en dedans vers le VIII (malformation de Mondini).
Les hypoacousies de perception existant dès la naissance sont des atteintes congénitales. Certaines, mais pas toutes, sont héréditaires, c’est-à-dire génétiques, et les autres sont la conséquence de complications de la grossesse. La plupart sont dues à des lésions de la cochlée. Il peut y avoir destruction des structures vasculaires membraneuses qui soutiennent l’organe de Corti (malformation de Scheibe) ou perte d’éléments nerveux en partant des cellules ciliées et en allant en dedans vers le VIII (malformation de Mondini).
Il est important de découvrir les hypoacousies congénitales le plus tôt possible afin de tout faire pour les compenser et d’adapter une prothèse auditive si c’est indiqué. La perception de la parole, et plus tôt elle est rétablie mieux c’est, joue un rôle capital dans le développement des centres auditifs du cerveau. Il existe une analogie avec l’œil : si chez un enfant atteint de strabisme on ne masque pas l’œil sain pour faire travailler l’œil non dominant, le cortex visuel du cerveau du côté correspondant ne se développe pas. Il en va de même pour le cortex auditif.
Les hypoacousies congénitales sont souvent suspectées avant l’âge d’un an. Malheureusement, beaucoup ne sont découvertes que beaucoup plus tard, Habituellement, la mère ou un autre membre de la famille sont les premiers à remarquer un problème. Il est alors important pour le clinicien de prendre en considération les inquiétudes de la famille et d’aller jusqu’au bout de l’évaluation audiométrique. Un audiologiste expérimenté peut se faire une bonne idée sur la possibilité d’une hypoacousie à l’aide de tests audiométriques de base dès la petite enfance.
Des informations diagnostiques d’excellente qualité peuvent être tirées des PEA. Ce test mesure les réponses auditives chez un nourrisson sous sédatif et les caractéristiques des courbes peuvent même indiquer si le problème se situe dans l’oreille moyenne, l’oreille interne ou le huitième nerf crânien. Les otoémissions acoustiques provoquées (OEAP) sont aussi un test objectil qui identifie des sons produits par les cellules ciliées externes saines sous l’effet d’un stimulus. De plus en plus d’unités de néonatologie hospitalières testent tous les nouveau-nés à l’aide des OEAP ou des PEA.
Les facteurs de risque prédominants d’atteinte auditive congénitale sont :
- les antécédents familiaux d’hypoacousie héréditaire ;
- la prématurité avec un poids de naissance inférieur à environ 1,5 kg ;
- l’atteinte de la mère par le virus de la rubéole pendant la grossesse ;
- l’existence de malformations maxillofaciales ;
- un ictère du nouveau-né avec un niveau de bilirubine supérieur à 12 mg/dl ;
- une infection néonatale, surtout en cas de méningite ;
- un accouchement aux forceps avec lésion de l’os temporal.
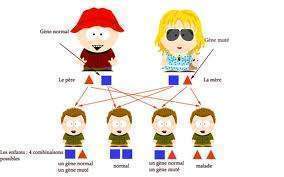
L’hypoacousie héréditaire ou familiale peut certes être présente à la naissance mais peut aussi se développer pendant l’enfance ou l’adolescence. Environ 20 p. 100 sont rapportées comme étant autosomiques dominantes, et 80 p. 100 comme récessives. L’âge de l’installation, la sévérité et l’aspect audiométrique de la perte varient considérablement parce que de nombreux sites génétiques peuvent être en cause. Un aspect souvent rencontré est celui de courbe en cuillère avec une chute nette sur les fréquences moyennes .
La plupart de ces pertes ne sont pas incluses dans des syndromes associant d’autres organes, mais un certain nombre le sont. Tout à fait étonnant, le syndrome de Waardenburg, autosomique dominant, associe des canthi internes des yeux anormalement écartés et une mèche de cheveux blancs. Un autre, le syndrome d’Usher, atteinte récessive, comprend aussi un problème oculaire, une rétinite pigmentaire. La sévérité de l’atteinte auditive dans ces deux syndromes est variable. Dans la plupart des atteintes familiales, l’hypoacousie est bilatérale. Un bilan génétique peut désormais localiser et typer le gène en cause.
Il y a des syndromes d’hypoacousie congénitale qui ne sont pas dus à une atteinte cochléaire mais intéressent en fait le nerf et les voies auditives plus centrales. La neuropathie auditive correspond à une incapacité du nerf auditif à envoyer un signal synchrone de bonne qualité vers le tronc çérébral. Ces enfants ont des cochlées intactes avec des OEAP normales mais des PEA anormaux. Ils peuvent percevoir des sons de toutes les fréquences mais ne peuvent pas comprendre ou acquérir la parole. L’ictère néonatal est à l’origine de certains cas mais pas de tous. Un certain nombre de sujets affectés ont d’autres neuropathies périphériques, et il y a probablement des facteurs génétiques.
Certains enfants ont des problèmes avec le traitement central du signal auditif, un dysfonctionnement situé quelque part entre le tronc cérébral et le cortex. En général, ils entendent toutes les fréquences normalement et ont une bonne discrimination vocale dans le silence des conditions du test, mais ont des performances médiocres quand il y a un bruit de fond ou des sources de distraction. Leurs problèmes sont moins graves que ceux des sujets atteints de neuropathie auditive et sont souvent découverts à l’occasion de troubles de l’apprentissage à l’école. Ils peuvent alors être mis en évidence par des tests audiométriques spécialisés.
Résumé du traitement
Il faut insister avec force sur l’importance de la détection précoce des hypo acousies congénitales. De plus en plus, les unités de néonatalogie hospitalière réalisent des dépistages systématiques par OEAP ou PEA chez les nouveau-nés. Il n’en est pas moins vrai que le généraliste doit prendre au sérieux les inquiétudes d’un membre de la famille, quel qu’il soit, qui sus pecte une hypoacousie et aller jusqu’au bilan audiométrique et ORL. Les traitements des atteintes discutées plus haut sont adaptés en fonction du type d’atteinte. La plupart des atteintes cochléaires, de légères à sévères, sont mieux compensées par un appareillage précoce. Les problèmes de traitement central de l’information auditive répondent bien à l’usage de dispositifs d’écoute à modulation de fréquence dans les situations d’apprentissage. Cela implique l’utilisation d’un microphone pour l’enseignant et le port d’un écouteur pour l’élève, pour éliminer le bruit de fond. La neuropathie auditive est rarement améliorée par les prothèses mais plutôt par les indications visuelles (cued speech, lecture sur les lèvres ou langage des signes) et peut-être par l’implant cochléaire, un dispositif électronique implanté chirurgicalement dans l’oreille interne par un chirurgien de l’oreille très spécialisé. Le patient atteint de surdité profonde a, lui aussi, le choix entre ces deux dernières options. Ces traitements ne sont concevables qu’au sein d’équipes dévouées associant audiologistes, orthophonistes et parfois chirurgiens spécialisés. On ne les mentionne ici que brièvement, le lecteur intéressé peut trouver ailleurs une information plus complète.